%matplotlib inline
# these two lines are only necessary to make the jupyter notebooks run on binder
import sys
sys.path.insert(0, "../..")
# We load the AbsorptionSpectrum class from aimstools
from aimstools.dielectric_function import AbsorptionSpectrum as ABS
from aimstools.preparation.utilities import monkhorstpack2kptdensity as m2d
# We load all tests in the directory "absorption" to check k-point convergence and omega convergence
from pathlib import Path
k_tests = list(Path().cwd().joinpath("absorption_spectrum/k_test").glob("k*"))
omega_tests = list(Path().cwd().joinpath("absorption_spectrum/omega_test").glob("omega*"))
# we initialize the AbsorptionSpectrum class for the k-tests
k_test = [ABS(i) for i in k_tests]
# we sort them by increasing k point densities
k_densities = sorted([m2d(j.structure, j.control.k_grid) for j in k_test])
k_test = [j[0] for j in (sorted(zip(k_test, k_densities), key=lambda x: x[1]))]
# we initialize the AbsorptionSpectrum class for the omega-tests
o_test = [ABS(i) for i in omega_tests]
# we sort them by increasing increasing omega value
omegas = sorted([int(str(j.parts[-1]).split("_")[1]) for j in omega_tests])
o_test = [j[0] for j in (sorted(zip(o_test, omegas), key=lambda x: x[1]))]
import matplotlib.pyplot as plt
import numpy as np
cmap = plt.cm.get_cmap("tab20_r")
colors = [cmap(c) for c in np.linspace(0, 1, len(k_test))]
fig, axes = plt.subplots(1,2,figsize=(12,6))
for i, n in enumerate(k_test):
n.plot(axes=axes[0], color=colors[i], component="total", label=str(k_densities[i]), alpha=0.8)
for i, m in enumerate(o_test):
m.plot(axes=axes[1], color=colors[i], component="total", label=str(omegas[i]), alpha=0.8)
axes[0].set_xlim(600, 300)
axes[1].set_xlim(600, 300)
axes[0].set_title(r"$k$-point density dependency")
axes[1].set_title(r"$\omega_{max}$ dependency")
plt.show()
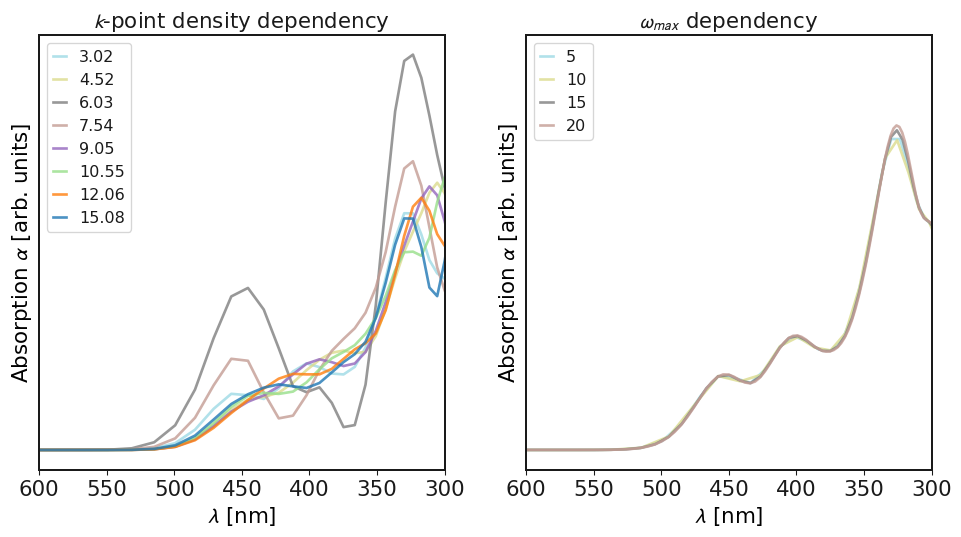
# As yo can see, the dependency on the k-point density is very large and needs to be checked carefully.
# Even for semiconductors, it seems like one needs 15-20 points/Angström.
# Regarding omega, positive values above zero in the range of 5-15 seem fine, depending on the cost of the calculation and the band structure.